Prader-Willi sindromas
Clemensas Gödelis yra laisvai samdomas darbuotojas „houseofgoldhealthproducts“ medicinos komandoje.
Daugiau apie „houseofgoldhealthproducts“ ekspertus Visą „houseofgoldhealthproducts“ turinį tikrina medicinos žurnalistai.Prader-Willi sindromas (PWS) yra įgimto genetinės medžiagos defekto rezultatas. Pažeisti kūdikiai yra trumpi, psichiškai nepakankamai išvystyti ir raumeningi. Ankstyvoje vaikystėje jiems pasireiškia nepasotinamas alkis, dėl kurio atsiranda ryškus nutukimas. Gautos ligos turi būti gydomos gydant įvairias medicinos specialybes. Skaitykite daugiau apie Prader-Willi sindromo simptomus, diagnozę ir gydymą čia!
Šios ligos TLK kodai: TLK kodai yra tarptautiniu mastu pripažinti medicininių diagnozių kodai. Jų galima rasti, pavyzdžiui, gydytojo laiškuose arba nedarbingumo pažymėjimuose. Q87
Prader-Willi sindromas: aprašymas
Prader-Willi sindromą (klaidingai taip pat Willi-Prader sindromą) 1956 metais pirmą kartą aprašė pediatrai Andrea Prader, Alexis Labhart ir Heinrich Willi. Maždaug vienas iš 20 000 naujagimių kenčia nuo Prader-Willi sindromo. Priežastis yra genetiškai sukeltas hipotalamo disfunkcija, svarbus smegenų perjungimo centras. Prader-Willi sindromo pasireiškimas gali būti labai įvairus ir sudėtingas.
Prader-Willi sindromas: simptomai
Paveikti vaisiai pastebimi dar prieš gimdymą. Jie pastebimai mažai juda gimdoje. Širdies ritmas yra mažesnis nei normalus. Gimimo metu vaisiai, turintys Prader-Willi sindromą, dažniau būna nenormalioje padėtyje motinos kūne. Kūdikiams reikia daug paramos gimdymo metu ir po jo.
Iškart po gimimo paveikti naujagimiai išsiskiria dėl sėdimo gyvenimo būdo, (raumenų) silpnumo ir mažo gimimo svorio. Tipiškas riksmas po gimdymo taip pat gali nebūti arba būti silpnas. Dėl ryškaus silpnumo ir dėl to atsiradusių čiulpimo bei rijimo sutrikimų kūdikiams sunku gerti.
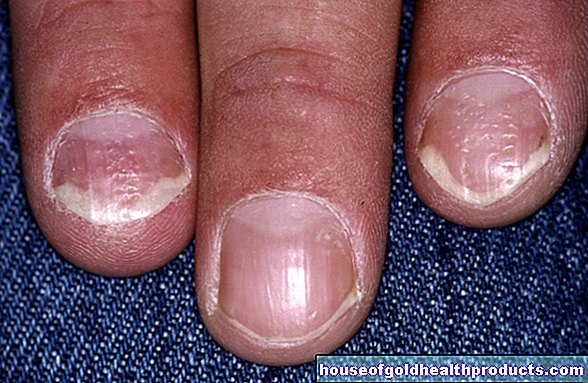
Kūdikiai, turintys Prader-Willi sindromą, dažnai turi tam tikrų išorinių savybių. Siauram veidui būdingos migdolo formos akys ir maža burna su plona viršutine lūpa. Kaukolė dažnai yra ilga (dolidochocefalija), o rankos ir kojos yra mažos. Stuburas gali būti sulenktas į S formą (skoliozė). Viso kūno kaulų medžiaga rodo rentgeno vaizdo pažeidimus ir defektus (osteoporozę / osteopeniją). Gali sumažėti odos, plaukų ir tinklainės pigmentacija. Taip pat galimi regos sutrikimai ir akių vingiuotumas (strabizmas). Kapšelis yra mažas ir dažnai tuščias (nenusileidusios sėklidės). Apskritai sergančių vaikų raida vėluoja.
Pirmųjų gyvenimo metų pabaigoje raumenų silpnumas šiek tiek pagerėja. Tačiau visada yra bent silpnas silpnumas. Įprasta veikla gali greitai tapti varginanti ir varginanti pacientams, sergantiems Praderio-Willi sindromu. Nepaisant to, vaikams taip pat patinka tipiška vaikystės veikla. Vaikystėje augimas žymiai sumažėja.
Neribotas maisto vartojimas
Ligai progresuojant, paveikti vaikai valgo vis daugiau (hiperfagija) - nesijaučiantys sotūs. Maistas kaupiamas ir net sunkiais atvejais pavogiamas. Vaikams labai sunku kontroliuoti savo mitybos įpročius. Todėl jiems išsivysto ryškus nutukimas (nutukimas). Jie ypač greitai priauga svorio, nes yra didelis neatitikimas tarp didelio kalorijų kiekio ir mažo energijos suvartojimo.
Antsvoris sukelia tipiškas antrines ligas: širdis ir plaučiai kenčia nuo nutukimo. Ketvirtadalis visų nukentėjusiųjų susirgo cukriniu diabetu iki 20 metų. Miego sutrikimai, venų ligos (tromboflebitas) ir vandens susilaikymas taip pat yra daugybė galimų pasekmių. Ligos eigoje gali atsirasti miego sutrikimų. Be pasikartojančių kvėpavimo pauzių, galimi dienos ir nakties ritmo ar gilaus miego sutrikimai. Paveiktiems vaikams sunku mokytis.
Sutrinka brendimo raida
Tipiškas augimo šuolis brendimo metu yra mažas. Sergantys vaikai paprastai neauga aukštesni nei 140–160 centimetrų. Nors brendimas gali prasidėti per anksti (priešlaikinis adrenarche), daugeliu atvejų brendimo vystymasis niekada nesibaigia. Paprastai nukentėjusieji išlieka sterilūs. Berniukams varpos ir ypač sėklidės lieka mažos. Ant kapšelio nėra pigmentacijos ir raukšlių. Gali nebūti balso pertraukos. Mergaitėms lytinės lūpos ir klitoris lieka nepakankamai išvystyti. Pirmasis kraujavimas iš menstruacijų nepasireiškia nei anksčiau, nei vėlai, kartais tik nuo 30 iki 40 metų.
Psichikos ir psichikos vystymasis
Prader-Willi sindromo atveju sutrinka tiek psichinis, tiek psichomotorinis vystymasis. Vaiko raidos etapai dažnai pasiekiami vėliau nei sveikų bendraamžių. Kalbinis ir motorinis vystymasis kartais užtrunka dvigubai ilgiau nei sveikų bendraamžių.
Vidutinis intelekto koeficientas (IQ) yra nuo 60 iki 70, taigi gerokai žemiau normos. Fizinis silpnumas gali apsunkinti kalbėjimą. Kalbos vystymasis vėluoja ir kalbos supratimas sutrinka, priklausomai nuo žemo intelekto koeficiento. Maždaug 40 procentų ligonių yra ant intelekto negalios slenksčio. Nepriklausomai nuo intelekto koeficiento, taip pat atsiranda mokymosi sutrikimų, tokių kaip aritmetiniai sunkumai. Mokyklos rezultatai dažniausiai yra žemesni nei vidutiniai.
Gali būti pastebimas ir emocijų vystymasis, ir elgesys. Paveikti žmonės kartais apibūdinami kaip užsispyrę ir greitai nusiteikę. Psichikos sutrikimai gali atsirasti jau ankstyvoje vaikystėje: aprašomos vadinamojo opozicinio elgesio formos, taip pat nelankstus ir turintis elgesys. Įprasti procesai gali būti sunkūs. Kartais procesus reikia kartoti priverstinai. Autistiniai bruožai pasireiškia maždaug 25 proc. Dėmesio deficito sutrikimas (ADD) taip pat yra dažnas reiškinys.
Simptomai paprastai didėja su amžiumi ir antsvoriu. Tačiau vyresnio amžiaus žmonėms Prader-Willi sindromo simptomai gali lengvai atsinaujinti. Maždaug dešimt procentų kenčia nuo psichozės. Be to, epilepsija ir „priklausomybės nuo miego“ (narkolepsijos) formos yra susijusios su Praderio-Willi sindromu.
Prader-Willi sindromas: priežastys ir rizikos veiksniai
Manoma, kad Prader-Willi sindromo priežastis yra dalies vidurinės smegenų, žinomos kaip pagumburis, disfunkcija. Be kita ko, tai lemia svarbaus augimo hormono trūkumą. Sutrikimą sukelia maždaug trys ketvirtadaliai atvejų dėl to, kad 15 chromosomoje nėra genų segmento (15q11-q13). Praderio-Willio sindromo atveju ypač svarbus tėvo chromosomos kopijos defektas (70–75 proc.) ), kad būtų tik viena geno kopija. Kita galimybė yra ta, kad abu dvigubo chromosomų rinkinio genai yra tik iš motinos (vienpusė disomija, 25–30 proc.). Vadinamasis „atspaudo defektas“ yra retesnis (vienas procentas). Terminas „įspaudas“ apibūdina tai, kad genai skaitomi priklausomai nuo jų kilmės (motinos ar tėvo pusėje).
Daugeliu atvejų sutrikimas nėra paveldimas. Paprastai jis vystosi tik vystantis lytinėms ląstelėms arba po apvaisinimo. Tačiau, kita vertus, gali būti, kad esamos genų mutacijos (dažniausiai vadinamosios subalansuotos translokacijos) taip pat sukelia Praderio-Willi sindromą. Tokiais atvejais padidėja paveldėjimo rizika.
Prader-Willi sindromas: tyrimai ir diagnozė
Visi naujagimiai, turintys nuolatinį ir nepaaiškinamą silpnumą, turi būti ištirti dėl PWS. Net naujagimių gydytojas (neonatologas) ar pediatras (pediatras) dėl vaiko elgesio įtaria Praderio-Willi sindromą po gimdymo. Jokie vadinamieji nuspėjamieji testai neatliekami be šio sindromo buvimo įrodymų. Tačiau visiškai įmanoma diagnozuoti Prader-Willi sindromą dar prieš gimdymą.
Fizinės ir techninės apžiūros
Daugeliu atvejų fizinis patikrinimas jau sukelia didelį įtarimą dėl Praderio-Willi sindromo (Holms Criteria 1993, 2001). Pagrindinė Prader-Willi sindromo savybė yra ryškus silpnumas, kuris ypač išryškėja geriant. Išvaizda taip pat suteikia užuominų. Paprastai aptinkami refleksai yra silpnai išreikšti.
Išmatuojamas augimo hormono trūkumas kraujyje taip pat yra naudingas diagnostikai. Lytinių hormonų (estrogenų, testosterono, FSH, LH) taip pat dažniausiai sumažėja sergantiems. Tai lydi nepakankamas lytinių organų išsivystymas. Antinksčių žievės funkcija daugeliu atvejų sutrinka. Lytinių hormonų (androgenų) susidarymas taip pat gali prasidėti per anksti (ankstyvas adrenarche).
Smegenų bangų tyrimas (elektroencefalograma, EEG) taip pat gali būti įtartinas.
Genetinis tyrimas
Atliekami genetiniai tyrimai, patvirtinantys įtarimą dėl Prader-Willi sindromo. Pirmajame etape tiriamas esminio 15 chromosomos taško (15q11.2-q13, „SNRPN lokusas“) metilinimas. Fermentai gali surišti vadinamąsias metilo grupes prie DNR ir taip ją modifikuoti. Daugiau nei 99 proc. Atvejų šis tyrimas suteikia diagnozę. Priešingu atveju atliekamas kitas įprastas chromosomų pokyčių aptikimo metodas - fluorescencijos in situ hibridizacija (FISH).
Panašios ligos yra Martin Bell sindromas arba Angelmann sindromas. Martin Bell sindromo sutrikimas yra X chromosomoje (trapus X sindromas). Esant Angelmanno sindromui ir Praderio -Vilio sindromui, ta pati 15 chromosomos padėtis daugeliu atvejų ištrinama, tačiau Angelmanno sindromo atveju tik ta, kuri yra motinos chromosomoje.
Prader-Willi sindromas: gydymas
Praderio-Willi sindromo negalima išgydyti. Tačiau taikant griežtai vadovaujamą, palaikomąją terapiją, simptomus galima palengvinti. Pagrindiniai gydymo komponentai yra maisto kontrolė, pakaitinė hormonų terapija ir elgesio problemų gydymas.
maitinimas
Turi būti užtikrinta tinkama mityba ir pakankamas augimas, ypač jei raumenų silpnumas yra stiprus. Siekiant palengvinti maisto vartojimą, gali būti naudojami zondai arba specialūs dirbtiniai speneliai. Be to, turėtų būti sudarytas šėrimo planas su gerai dokumentuotu kalorijų suvartojimu ir svorio kontrole.
Tačiau augant vaikams išsivysto persivalgymo sutrikimas. Tada reikia laikytis plano su griežtu kalorijų apribojimu. Ne visada siekiama apriboti riebalus, nes riebalai yra svarbūs ragų vystymuisi. Dėl valgymo sutrikimo gali tekti griežtai valdyti maistą. Valgymo sutrikimas sergant Prader-Willi sindromu daro ypač rimtą įtaką ligos eigai, nes nukentėjusieji mažai juda, todėl yra didelis kalorijų suvartojimo ir poreikio neatitikimas. Labai svarbu aktyviai įtraukti tėvus ir pasiūlyti sergančiam vaikui tvirtą struktūrą.
Tuo pačiu metu būtina užtikrinti pakankamą vitaminų ir mineralų kiekį.Esant Prader-Willi sindromui, dažnai pasitaiko kaulų apykaitos sutrikimų, kurių galima išvengti vartojant vitamino D ir kalcio. Skeleto, ypač stuburo, vystymąsi reikia reguliariai stebėti.
Sergančio vaiko veikla ir motorinė raida reguliariai tikrinama ir prireikus terapiškai palaikoma fizioterapija ar panašiais metodais.
Augimo hormonas
Nuo dvejų metų augimo hormonas HGH taip pat gali būti skiriamas kaip vaistas. Ši terapija turi būti nutraukta, kai kaulų augimo plokštelės yra uždarytos (rentgeno kontrolė). Hormonų terapija visada turi būti atidžiai stebima. Hormonų vartojimas teigiamai veikia kūno vystymąsi, tačiau šalutinis poveikis yra pėdų edema, stuburo kreivumo pablogėjimas (skoliozė) arba kaukolės slėgio padidėjimas (pseudotumor cerebri). Gydymo pradžioje gali atsirasti kvėpavimo sutrikimų. Todėl gydymo pradžioje svarbu stebėti miegą. HGH hormonų terapijos stebėjimas taip pat apima reguliarius skydliaukės ir augimo faktoriaus IGF-1 lygio matavimus kraujyje.
Lytinio brendimo atveju lytiniai hormonai gali būti skiriami kaip depo injekcijos, hormonų pleistrai ar gelis. Tai gali pagerinti elgesio problemas. Estrogenai taip pat padeda kurti kaulus, tačiau jie taip pat turi nemažai šalutinių poveikių.
Psichologinė parama
Nukentėjęs vaikas turėtų gauti pagalbą, kad padėtų ugdyti elgesį ir pasiekti vystymosi etapus. Ypač socialinius įgūdžius reikia lavinti sergant Praderio-Willi sindromu. Taip pat turėtų būti skatinamas bendravimas su bendraamžiais ir globėjais. Mokykloje gali prireikti individualios priežiūros. Jei reikia, gyvenimo ir darbo aplinka turi būti pritaikyta. Psichikos sutrikimams gali prireikti gydymo vaistais, pavyzdžiui, naudojant serotonino antagonistą. Intensyvios paramos tikslas - kuo geresnė nepriklausomybė.
Chirurgijos reikmenys
Bet kokį lūpų ir gomurio plyšį, kuris gali atsirasti jau gimus, chirurgai gali gydyti ankstyvoje stadijoje. Akių pažeidimus, ypač žvilgsnio padėtį, taip pat galima gydyti chirurginiu būdu, kad būtų išvengta regos sutrikimų. Pirmiausia gali padėti laikinai uždengti sveiką akį.
Dėl nepakankamo lytinių organų išsivystymo gali prireikti operacijos, kad sėklidė būtų perkelta iš apatinės pilvo dalies į kapšelį. Beta-hCG (žmogaus chorioninis gonadotropinas) gali padidinti kapšelį ir leisti sėklidei nuskęsti.
Skeleto pokyčiai Prader-Willi sindromo atveju taip pat gali būti gydomi chirurginiu būdu. S stuburo S padėtis (skoliozė) sunkiais atvejais reikalauja chirurginės korekcijos, tačiau dažniausiai gydoma be operacijos (pavyzdžiui, su korsetu).
Prader-Willi sindromas: ligos eiga ir prognozė
Ankstyviausia įmanoma „Prader-Willi sindromo“ diagnozė gali turėti teigiamą poveikį ilgalaikei prognozei. Teigiama įtaka (valgymo) elgesiui ir galimas augimo hormonų vartojimas gali pagerinti nukentėjusio vaiko gyvenimo kokybę.
Po diagnozės reguliariai reikia tikrinti valgymo elgesį, svorį ir augimą. Tikrinamas vystymasis, funkcija, elgesys ir psichikos sutrikimai. Labai svarbu atidžiai stebėti ir rūpintis specialistu, kuris gali koordinuoti ir užtikrinti tarpdisciplininę priežiūrą.
Sergantiems žmonėms padidėja rizika pakartotinai užsikrėsti infekcijomis. Operacijų metu reikia būti ypač atsargiems: po anestezijos pabudimo fazė dažnai trunka ilgiau ir padidėja kvėpavimo sutrikimų rizika.
Tačiau didžiausia problema yra didėjantis nutukimas. Gyvenimo metu vyrauja atsiradusios komplikacijos. Mirtingumas taip pat padidėja dėl komplikacijų. Todėl padidėjęs Prader-Willi sindromo mirtingumas pirmiausia atsiranda dėl širdies, kraujagyslių ar plaučių ligų.
Paveldėjimo rizika yra maža. Daugeliu atvejų genetiniai pokyčiai yra spontaniški ir nėra paveldimi. Tėvų rizika susilaukti antro vaiko su Praderio-Willi sindromu yra maža. Nukentėjusieji dažnai lieka bevaikiai.
Tik retais atvejais Praderio-Willio sindromą sukelia tėvų chromosomų rinkinio genetinės informacijos pokyčiai, kurie yra paveldimi ir taip pat lemia didesnę paveldėjimo tikimybę. Jei vaikas serga Praderio-Vilio sindromu, rekomenduojame tėvams, norintiems turėti vaikų, kreiptis patarimo į žmogaus genetikus.
Žymos: sporto fitnesas odos priežiūra kūdikis kūdikis