Fenilketonurija
Marianas Grosseris Miunchene studijavo žmonių mediciną. Be to, daugeliu dalykų besidomintis gydytojas išdrįso padaryti įdomių aplinkkelių: studijuoti filosofiją ir meno istoriją, dirbti radijuje ir, galiausiai, „Netdoctor“.
Daugiau apie „houseofgoldhealthproducts“ ekspertus Visą „houseofgoldhealthproducts“ turinį tikrina medicinos žurnalistai.Fenilketonurija (PKU) yra įgimta, paveldima baltymų apykaitos liga. Jis apsaugo nuo aminorūgšties fenilalanino skilimo. Tai kaupiasi organizme ir sutrikdo vaiko smegenų vystymąsi. Jei negydoma, fenilketonurija sukelia sunkią intelekto negalią. Tačiau laiku gydant, pacientai gali gyventi normalų gyvenimą. Sužinokite viską apie fenilketonuriją čia!
Šios ligos TLK kodai: TLK kodai yra tarptautiniu mastu pripažinti medicininių diagnozių kodai. Jų galima rasti, pavyzdžiui, gydytojo laiškuose arba nedarbingumo pažymėjimuose. E70
Fenilketonurija: aprašymas
Fenilketonurija (PKU) yra paveldima medžiagų apykaitos liga, kuri egzistuoja nuo gimimo ir trukdo skaidyti nepakeičiamą aminorūgštį fenilalaniną. Aminorūgštys yra pagrindiniai baltymų elementai, taigi ir gyvybiškai svarbūs medžiagų apykaitos komponentai. Kai kurie iš jų gali patekti į organizmą tik per maistą, organizmas negali jų pats pasigaminti. Tokios amino rūgštys vadinamos nepakeičiamomis.
Kas atsitinka su fenilketonurija?
Paprastai aminorūgštys yra subalansuotos tarp absorbcijos / kaupimosi ir suskaidymo, todėl visada yra tiek, kiek reikia organizmui. Įvairių amino rūgščių atveju trūkumas ar perteklius gali padaryti didelę žalą ir sukelti įvairius simptomus.
Vadinamajame klasikiniame PKU fenilalanino hidroksilazės (PAH) poveikis yra ribotas arba jo visai nėra. Dėl PAH trūkumo fenilalaninas vis dažniau kaupiasi organizme. Per didelė fenilalanino koncentracija labai sutrikdo smegenų vystymąsi ir ankstyvoje stadijoje jauniems pacientams sukelia intelekto sutrikimus.
Kadangi sergant šia liga normalus fenilalanino skilimas neįmanomas, susidaro kiti skilimo produktai, vadinamieji fenilketonai. Jie išsiskiria su šlapimu ir yra atsakingi už ligos pavadinimą.
Netipinė fenilketonurija
Net esant netipinėms fenilketonurijos formoms, sutrinka fenilalanino skilimas. Tačiau priežastis nėra PAH defektas. Vietoj to, kofermento, tetrahidrobiopterino (BH4), funkcija yra ribota. Jis netiesiogiai dalyvauja skaldant fenilalaniną, nes PAH reikia BH4, kad fenilalaninas taptų tirozinu.
Kadangi BH4 taip pat svarbus pasiuntinių dopamino ir serotonino gamybai, netipinė fenilketonurija su BH4 trūkumu paprastai yra sudėtingesnė nei klasikinė forma.
Kam veikia fenilketonurija?
PKU yra viena iš labiausiai paplitusių įgimtų medžiagų apykaitos ligų. Skaičiuojama, kad maždaug vienas iš 7 000 naujagimių visame pasaulyje jį išsivystys, nesiskiria tarp mergaičių ir berniukų. Kadangi tai yra paveldima liga, dažnai serga keli šeimos nariai.
Feniketonurija: simptomai
Iš pradžių kūdikiams, sergantiems fenilketonurija, nėra jokių ligos simptomų. Pirmosios problemos atsiranda tik ketvirtą – šeštą gyvenimo mėnesį, jei liga dar nebuvo pripažinta ir gydoma. Visų pirma, sutrikęs smegenų brendimas laikui bėgant sukelia didelių komplikacijų. Neapdoroto PKU simptomai yra šie:
- stiprus psichikos sutrikimas. Smegenų pažeidimas progresuoja brendimo metu ir stagnuoja. Tada nukentėję vaikai paprastai yra sunkiai protiškai neįgalūs.
- Traukuliai (traukuliai) (epilepsija). Dėl žalos smegenų nervinės ląstelės yra ypač jautrios ir pernelyg jaudinančios. To pasekmė yra dažni epilepsijos priepuoliai.
- motoriniai sutrikimai. Ne tik smegenų ląstelės, bet ir paciento raumenys gali būti pernelyg susijaudinę. Štai kodėl jis dažnai tampa įtemptas (spazmas), dėl kurio atsiranda įvairių judesių sutrikimų.
- Elgesio sutrikimai. Kai kurie vaikai, sergantys fenilketonurija, yra hiperaktyvūs ir neįprastai agresyvūs, taip pat dažniau pasitaiko pykčio priepuolių.
- maža galva (mikrocefalija). Kadangi paciento smegenys netinkamai vystosi, galvos augimas taip pat atsilieka. Maža galvos apimtis, lyginant su bendraamžiais, ypač pastebima vyresniems vaikams.
- pastebimas kvapas. PKU gamina tam tikrus fenilalanino skilimo produktus, kurie kvepia panašiai kaip pelių ekskrementai. Šios medžiagos daugiausia išsiskiria su šlapimu, bet taip pat iš dalies per odą.
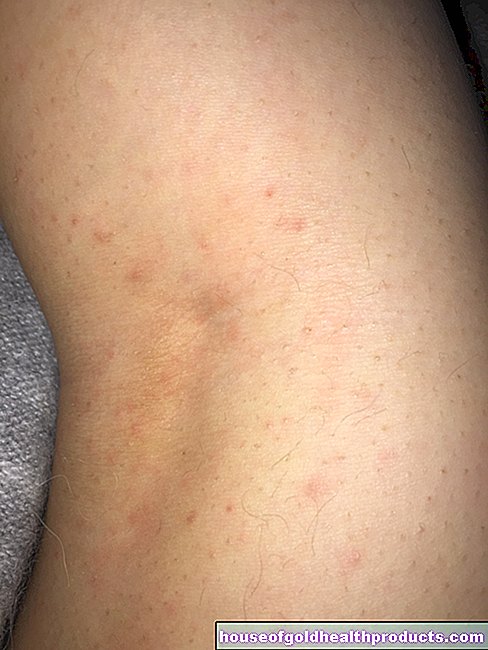
- į egzemą panašūs odos pokyčiai
Kadangi sergant fenilketonurija taip pat sutrinka pigmento melanino gamyba, daugelis sergančiųjų turi labai šviesią, jautrią saulei odą ir šviesiai šviesius plaukus. Akių rainelė taip pat yra nuo šviesiai mėlynos iki skaidrios ir leidžia rausvam dugnui šviesti.
PKU simptomai kiekvienam asmeniui skiriasi. Pagrindinė to priežastis yra ta, kad fenilalanino hidroksilazės (PAH) aktyvumas kiekvienam pacientui yra ribojamas skirtingai. Kai kurie vis dar turi tam tikrą liekamąjį aktyvumą, todėl organizme susikaupia mažiau fenilalanino. Kiti nerodo jokio fermento aktyvumo - liga atitinkamai progresuoja greičiau ir rimčiau.
Fenilketonurija: priežastys ir rizikos veiksniai
Fenilketonurija yra paveldima liga. Dabar žinoma daugybė genetinių mutacijų, dėl kurių atsiranda PAH defektas. Mutacijos tipas nustato, kiek ribojamas fenilalanino skilimas.
PKU paveldima recesyviai, o tai reiškia, kad žmogus gali būti pakeisto geno nešiotojas, nesivystydamas ligai. Panašiai žmonės, sergantys fenilketonurija, gali susilaukti sveikų vaikų.
Tik tuo atveju, jei abu tėvai turi mutacijų savo genetinėje sudėtyje, yra tam tikra tikimybė, kad jų palikuonys susirgs fenilketonurija. Jei tėvai yra ne tik genų nešiotojai, bet ir patys turi PKU, visi vaikai kartu taip pat jį vystys.
Fenilketonurija: tyrimai ir diagnozė
Kadangi rimtų fenilketonurijos pasekmių galima išvengti pradėjus gydymą laiku, ypač svarbu ligą atrasti kuo anksčiau. Vokietijoje trečią dieną po gimimo vaikai tiriami dėl įvairių įgimtų ligų, įskaitant PKU.
Tandeminė masių spektrometrija
Daugelis įgimtų medžiagų apykaitos sutrikimų dabar diagnozuojami vadinamosios tandeminės masės spektrometrijos pagalba. Tai leidžia greitai ir lengvai ištirti naujagimio kraują. Be fenilketonurijos, gydytojai per kelias minutes gali aptikti daugiau nei 20 kitų ligų.
Guthrie testas
Guthrie testas, pavadintas jo išradėjo vardu, taip pat leidžia diagnozuoti PKU. Norėdami tai padaryti, iš vaiko kulno paimamas nedidelis kiekis kraujo ir uždedamas ant filtro popieriaus. Tada laboratorijoje galite nustatyti, ar fenilalanino koncentracija yra padidėjusi.
Guthrie testas buvo pradėtas teikti praėjusio amžiaus septintajame dešimtmetyje ir jau seniai yra standartinis fenilketonurijos diagnozavimo metodas. Tačiau, palyginti su tandemo masės spektrometrija, ji turi trūkumų. Guthrie testas duoda rezultatą tik po penkių dienų, kuris taip pat yra linkęs į klaidas. Pavyzdžiui, tokie veiksniai, kaip vaiko mityba ar galimas gydymas antibiotikais, klastoja rezultatus. Kai kuriose šalyse Guthrie testas vis dar naudojamas, Vokietijoje jis dažniausiai nebevartojamas.
Kiti bandymai
Jei naujagimių tikrinimas atskleidžia įtarimą dėl fenilketonurijos, atliekamas tolesnis tyrimas patvirtinimui. Taip pat galima nustatyti tikslią fenilalanino koncentraciją kraujyje.
Galiausiai svarbu atskirti, ar tai tipiškas (klasikinis), ar netipinis PKU. Tam taip pat galimi specialūs testai, pavyzdžiui, tetrahidrobiopterino testas nepalankiausiomis sąlygomis. Skirtumas yra svarbus, nes netipinė fenilketonurija traktuojama kitaip nei klasikinė forma.
Amniono skysčio tyrimas
Fenilketonuriją galima diagnozuoti jau nėštumo metu (prenatalinė diagnozė). Norėdami tai padaryti, iš motinos vaisiaus maišelio paimamas nedidelis kiekis vaisiaus vandenų ir tiriamos jame esančios negimusio vaiko ląstelės. Tokiu būdu galima nustatyti bet kokius genetinius defektus, sukeliančius PKU.
Tačiau tikrinant naujagimius, fenilketonurija paprastai aptinkama pakankamai anksti, kad ją būtų galima gydyti. Kadangi vaisiaus vandenų tyrimas visada yra susijęs su tam tikra rizika, jo naudojimas PKU diagnozei paprastai nėra naudingas.
Fenilketonurija: gydymas
Yra tik vienas būdas kovoti su fenilalanino pertekliumi PKU: sergantys vaikai turi laikytis specialios dietos, kad su maistu galėtų gauti kuo mažiau fenilalanino. Jiems taip pat reikia tam tikrų maisto papildų, kurie pakeistų medžiagas, kurios sveikiems vaikams susidarytų iš fenilalanino.
Terapija turi prasidėti prieš pasireiškiant pirmiesiems raidos sutrikimo simptomams, t. Y. Per pirmuosius du gyvenimo mėnesius. Smegenų pažeidimo, kuris jau įvyko, negalima pakeisti.
Mitybos terapija su PKU dieta
Visi natūralūs baltymai sudaro apie penkis procentus amino rūgšties fenilalanino. Daugelyje maisto produktų jo yra daug daugiau, nei reikia organizmui. Tai nėra problema sveikam žmogui, nes jis suskaido ir pašalina perteklinį fenilalanino kiekį.
Kita vertus, pacientams, sergantiems fenilketonurija, dažniausiai reikia apsieiti be natūralių baltymų. Pramoniniu būdu pagaminti specialūs produktai pakeičia trūkstamus maisto komponentus. Viena vertus, norima užkirsti kelią fenilalanino pertekliui, kita vertus, pacientui turėtų būti tiekiamos medžiagos, kurių kitu atveju trūktų, pavyzdžiui, tirozinas, kuris susidaro iš fenilalanino.
Tačiau PKU dietos tikslas nėra visiškai sustabdyti fenilalanino absorbciją. Kadangi organizmui reikia tam tikro kiekio amino rūgščių svarbiems medžiagų apykaitos procesams. Tinkamos koncentracijos pasiekimas yra didelis iššūkis gydytojams ir pacientams ir reikalauja daug disciplinos.
Todėl dietos reikia laikytis visą gyvenimą, bet ypač griežtai iki šešerių metų. Kadangi smegenys iki šio amžiaus labai stipriai vystosi ir todėl yra ypač jautrios pažeidimams. Suaugusiesiems didelė fenilalanino koncentracija nesukelia smegenų pažeidimo, kaip ir vaikams. Tačiau jie gali sukelti kitus neurologinius skundus, tokius kaip prasta koncentracija ar sulėtėjusios reakcijos.
Dietinis fenilketonurijos gydymas turėtų prasidėti specializuotame medžiagų apykaitos ligų centre. Kadangi ne kiekvienoje klinikoje galima nurodyti tėvams laikytis dietos. Tai geriausia padaryti padedant dietos konsultacijoms, kurios parodo, kaip laikytis dietos ir reguliariai stebėti fenilalanino kiekį kraujyje.
Netipinės fenilketonurijos gydymas
Netipinėse PKU formose trūkstamas kofermentas BH4 ir tam tikros pasiuntinės medžiagos, tokios kaip dopaminas ir serotoninas, yra dirbtinai pakeičiamos. Kai kuriais atvejais pacientas taip pat turi laikytis dietos, kurioje mažai fenilalanino.
Fenilketonurija nėštumo metu
Jei esate nėščia ir pati sergate fenilketonurija, turėtumėte apsvarstyti šiuos dalykus:
- Ypač griežtai laikykitės dietos!
- Dažnai patikrinkite fenilalanino kiekį kraujyje. Kadangi negimusiam vaikui fenilalanino koncentracija yra maždaug dvigubai didesnė nei nėščiai moteriai. Be smegenų, jie taip pat gali pakenkti negimusio vaiko širdžiai ir akims. Vaiko skeleto apsigimimai taip pat gali atsirasti dėl didelio fenilalanino kiekio nėštumo metu.
- Kad būtų išvengta vaiko smegenų pažeidimo ankstyvuoju nėštumo laikotarpiu, moterys, sergančios fenilketonurija, turėtų atidžiai planuoti nėštumą ir nuo pat pradžių imtis atsargumo priemonių.
- Nėščioms moterims, sergančioms fenilketonurija, griežtai nesilaikoma dietos gali sukelti persileidimą (savaiminį abortą).
- Bet kuriuo atveju nėščios moterys, sergančios fenilketonurija, turėtų kreiptis į gydytoją patarimo ir patarimo.
Fenilketonurija: ligos eiga ir prognozė
Jei fenilketonurija diagnozuojama anksti, jei įmanoma, naujagimiui ir jei laikomasi specialios PKU dietos, prognozė paprastai būna gera. Vaikai dvasiškai vystosi ir jų vidutinė gyvenimo trukmė.
Tačiau negydomas smegenų pažeidimas sukelia sunkius psichinės raidos sutrikimus, kurių vėliau ištaisyti neįmanoma. Nukentėjusiųjų gyvenimo trukmė yra normali, tačiau jų intelekto koeficientas beveik visada yra gerokai mažesnis už normą.
Ypatingas atvejis yra reta netipinė fenilketonurijos forma, kai yra BH4 trūkumas. Šis fenilketonurijos variantas gali sukelti progresuojantį neurologinį pažeidimą ir stiprų mėšlungį, nepaisant dietos.
Žymos: kūdikis kūdikis rūkymas terapijos